Long QT Syndrome and Brugada Syndrome
Inherited (or acquired) channelopathies causing prolonged QT (LQTS) or RBBB-like ST elevation (Brugada) — both predispose to polymorphic VT and sudden cardiac death.
Also known as: LQTS, long QT syndrome, torsades de pointes, Brugada syndrome, channelopathy
Overview
Inherited channelopathies of cardiac repolarization producing characteristic ECG patterns and predisposing to malignant ventricular arrhythmias. Long QT syndrome (LQTS) features prolonged ventricular repolarization (corrected QT interval) and risk of torsades de pointes; acquired forms are commonly drug- or electrolyte-induced. Brugada syndrome features a coved-type ST elevation in V1-V2 with risk of polymorphic VT or ventricular fibrillation, often during sleep or febrile illness.
Epidemiology
LQTS: prevalence ~1 in 2,000; LQT1, LQT2, and LQT3 account for ~75% of genotyped cases. Brugada syndrome: prevalence ~5 per 10,000 in Europeans, much higher in Southeast Asia (lai tai, pokkuri, bangungut — historically a leading cause of sudden death in young men). Brugada is more common in males ~8-10:1.
Keep reading — start your free trial
You've read your 2 free diagnosis previews. Create your free account to unlock the full Long QT Syndrome and Brugada Syndrome outline — plus all 514 diagnoses, 5,500+ board-style questions, flashcards, and an AI tutor. Your 7-day free trial includes everything, and there's no credit card required.
Risk factors
- LQTS: family history of sudden death or unexplained syncope; congenital deafness (Jervell-Lange-Nielsen syndrome, LQT1 with autosomal recessive deafness)
- LQTS triggers: LQT1 — exertion (especially swimming); LQT2 — auditory stimuli or postpartum; LQT3 — sleep / bradycardia
- Acquired LQTS: methadone, fluoroquinolones (moxifloxacin, levofloxacin), macrolides (azithromycin, clarithromycin), antipsychotics (haloperidol, ziprasidone, quetiapine), antiemetics (ondansetron, droperidol), antiarrhythmics (sotalol, dofetilide, ibutilide, amiodarone), hypokalemia, hypomagnesemia, hypocalcemia
- Brugada: family history of unexplained sudden death; fever (key trigger); sodium channel blockers (flecainide, propafenone, certain antidepressants); cocaine; vagotonic states (sleep, large meals)
Pathophysiology
LQTS results from mutations affecting cardiac repolarization channels: KCNQ1 (LQT1, slow delayed rectifier potassium current IKs), KCNH2 (LQT2, rapid delayed rectifier IKr), and SCN5A (LQT3, late sodium current INa). Prolonged action potential predisposes to early afterdepolarizations and triggered activity, generating torsades de pointes. Acquired LQTS most often results from drugs blocking the IKr (hERG) channel. Brugada syndrome is associated with SCN5A loss-of-function mutations affecting the cardiac sodium channel, producing transmural voltage gradients in the right ventricular outflow tract that promote phase 2 reentry and polymorphic VT.
Clinical presentation
Symptoms
- Recurrent unexplained syncope, especially in childhood or young adulthood
- Documented or suspected torsades de pointes / polymorphic VT
- Sudden cardiac death (may be the first manifestation)
- Brugada: arrhythmic events often during sleep or fever; nocturnal agonal respiration
- Seizure (often a misdiagnosis of arrhythmic syncope)
- Family history of premature sudden death (<40 years)
Signs / physical exam
- Exam is generally normal between events
- Bradycardia in LQT3 patients (prolongs QT further)
- Congenital sensorineural deafness in Jervell-Lange-Nielsen syndrome (LQTS)
Classic findings
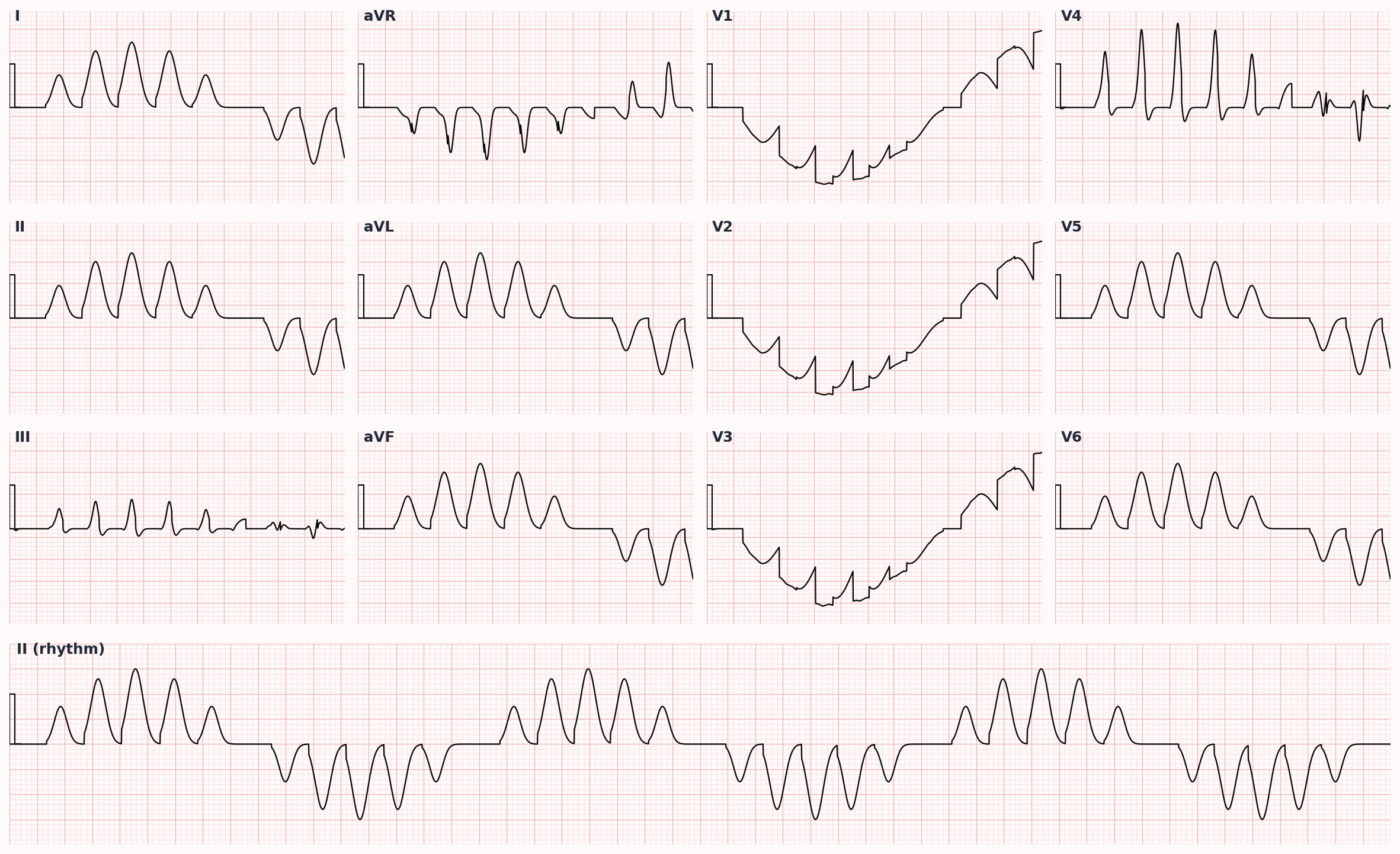
Adolescent or young adult with exertional syncope and prolonged QTc (LQT1); young male with nocturnal sudden death and coved ST elevation in V1-V2 (Brugada Type 1).
Differential diagnosis
- Acquired vs congenital LQTS — Medication review, electrolytes, family history; targeted genetic testing if congenital suspected
- Catecholaminergic polymorphic VT (CPVT) — Exercise- or emotion-induced bidirectional or polymorphic VT with normal resting ECG; RYR2 or CASQ2 mutations
- Short QT syndrome — QTc <340 ms with risk of AFib and sudden death; rare
- Early repolarization syndrome — J-point elevation in inferior or lateral leads; small additional sudden death risk; rarely needs ICD
- Right bundle branch block (true) — Brugada Type 1 ECG differs by coved ST elevation and inverted T wave in V1-V2; flecainide or ajmaline provocation if uncertain
- ARVC (arrhythmogenic right ventricular cardiomyopathy) — Epsilon waves, T-wave inversion V1-V3, structural RV disease on MRI
- Pulmonary embolism — Acute RV strain, S1Q3T3, but coved ST elevation V1-V2 is not the typical pattern
Diagnostic workup
Diagnostic criteria
Schwartz score for LQTS combines QTc duration, history of torsades, syncope (with/without stress), family history, and congenital deafness. Brugada syndrome is diagnosed by a spontaneous or drug-induced Type 1 pattern in ≥1 right precordial lead, plus clinical features per the 2013 HRS/EHRA/APHRS expert consensus.
Labs
- BMP including potassium, magnesium, calcium
- TSH
- Toxicology screen if drug-induced arrhythmia suspected
Imaging
- 12-lead ECG with manual QT measurement and Bazett or Fridericia correction (Bazett: QTc = QT / √RR). Normal QTc <450 ms in men and <460 ms in women; LQTS strongly suspected if QTc ≥480 ms or borderline (450-480) with clinical risk factors
- Brugada ECG patterns: Type 1 (coved-type ≥2 mm ST elevation followed by negative T wave in V1-V2) is diagnostic; Type 2 (saddleback) is suggestive but not diagnostic, may require provocation
- Exercise stress testing and ambulatory monitoring
- Echocardiography to exclude structural disease
- Sodium channel blocker challenge (procainamide, ajmaline, flecainide) under monitored conditions to unmask Brugada Type 1 ECG when suspicion is high but resting ECG is non-diagnostic
- Genetic testing (especially when ECG diagnostic): KCNQ1, KCNH2, SCN5A panels for LQTS; SCN5A for Brugada (positive in only ~20-30% of Brugada — clinical and ECG diagnosis remains primary)
Diagnostic algorithm
| Feature | Long QT Syndrome | Brugada Syndrome |
|---|---|---|
| Channel/gene | KCNQ1 (LQT1), KCNH2 (LQT2), SCN5A (LQT3) | SCN5A (loss-of-function) in 20-30% |
| ECG | Prolonged QTc (≥460-480 ms typical threshold) | Coved ≥2 mm ST elevation, V1-V2 (Type 1) |
| Trigger | LQT1 exercise/swim; LQT2 noise/postpartum; LQT3 sleep | Fever, sleep, vagotonic states, sodium channel blockers |
| Arrhythmia | Torsades de pointes (polymorphic VT) | Polymorphic VT / VF |
| Demographics | Children, young adults, both sexes | Adult males >> females, Southeast Asian heritage |
| Acute therapy | IV magnesium, correct electrolytes | Defibrillation, isoproterenol, quinidine |
| Long-term therapy | Beta-blocker (nadolol, propranolol) ± ICD ± LCSD | ICD for high-risk; quinidine adjunct |
Treatment
First-line
- LQTS — lifestyle: avoid QT-prolonging drugs (consult www.crediblemeds.org), maintain normal potassium/magnesium, avoid triggers based on genotype (e.g., competitive swimming in LQT1, sudden loud noises in LQT2)
- LQTS — beta-blocker (nadolol or propranolol preferred over selective agents) for all symptomatic patients and most asymptomatic patients with QTc ≥470 ms — first-line therapy; titrate to attenuate exercise heart rate response
- LQTS — ICD: aborted cardiac arrest, recurrent syncope on adequate beta-blockade, or very high-risk genotype/QTc combinations
- LQTS — left cardiac sympathetic denervation (LCSD) for high-risk patients with breakthrough events on beta-blocker
- Acute torsades de pointes: IV magnesium 2 g over 5-15 min (regardless of magnesium level), correct hypokalemia, withdraw offending agents, overdrive pacing or isoproterenol (if no contraindication and not LQTS-related) for pause-dependent torsades, defibrillate sustained VT/VF
Second-line / adjunct
- Brugada — avoid triggers: aggressive fever control (acetaminophen), avoid sodium channel blocker antiarrhythmics, certain psychotropics (some tricyclics, lithium), cocaine, excessive alcohol
- Brugada — ICD is the only therapy proven to prevent sudden cardiac death; indicated for cardiac arrest survivors, documented sustained VT, or spontaneous Type 1 ECG with syncope or family history of SCD
- Brugada — quinidine for ICD storm or for patients with high-risk features who are not ICD candidates
- Brugada — substrate ablation of the RV outflow tract epicardium for recurrent VT or refractory ICD shocks in expert centers
- Family screening with ECG ± genetic testing for first-degree relatives in both LQTS and Brugada
Complications
- Polymorphic VT, torsades de pointes, ventricular fibrillation
- Sudden cardiac death
- ICD shocks (appropriate and inappropriate), psychological impact, complications of device therapy
- Drug interactions in patients on chronic medications who must avoid QT-prolonging therapies (e.g., azithromycin, ondansetron)
PANCE pearls
- Always measure QTc manually — automated QT measurements are unreliable. Use the lead with the clearest T-wave end (often II or V5) and apply Bazett (or Fridericia at extreme heart rates).
- LQTS genotype-trigger associations: LQT1 = exertion / swimming; LQT2 = auditory stimuli / postpartum; LQT3 = rest / sleep.
- Acute torsades: IV magnesium FIRST, regardless of serum magnesium level; then correct potassium, remove offending drugs, and consider overdrive pacing for pause-dependent forms.
- Brugada Type 1 ECG (coved-type) is diagnostic; fever frequently unmasks the pattern — counsel patients to be aggressive with antipyretics.
- Family screening (ECG ± genetic testing) is essential in both LQTS and Brugada — first-degree relatives should be offered evaluation.
Images
References
- HRS/EHRA/APHRS 2013 — HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes (Priori et al., Heart Rhythm 2013)
- ACC/AHA/HRS 2017 — 2017 ACC/AHA/HRS Guideline for Management of Patients With Ventricular Arrhythmias and Prevention of Sudden Cardiac Death (Al-Khatib et al., JACC 2018)
- ESC 2022 — 2022 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death (Zeppenfeld et al., Eur Heart J 2022)
- CredibleMeds — QTdrugs lists, AzCERT (www.crediblemeds.org)
Practice Cardiovascular questions on FirstPassPA
Turn this outline into retention. 5,500+ board-style questions with an AI tutor that explains every answer — free to start, no card required.
Start studying free → Browse all 514 diagnosesEducational use only. This outline is a study aid for PA students and is not medical advice or a substitute for clinical judgment. FirstPassPA is an independent study tool and is not affiliated with, endorsed by, or sponsored by NCCPA or PAEA. PANCE® and PANRE® are registered trademarks of the National Commission on Certification of Physician Assistants; End of Rotation™ is a program of the Physician Assistant Education Association.